Saúde
Cientistas descobrem a genética por trás do vazamento de vasos sanguíneos cerebrais na síndrome de Rett
Ao demonstrar que o problema deriva de mutações genéticas que levam à superexpressão de um microRNA, o estudo dos pesquisadores do MIT aponta para um possível tratamento
Por
David Orenstein - 20/03/2026


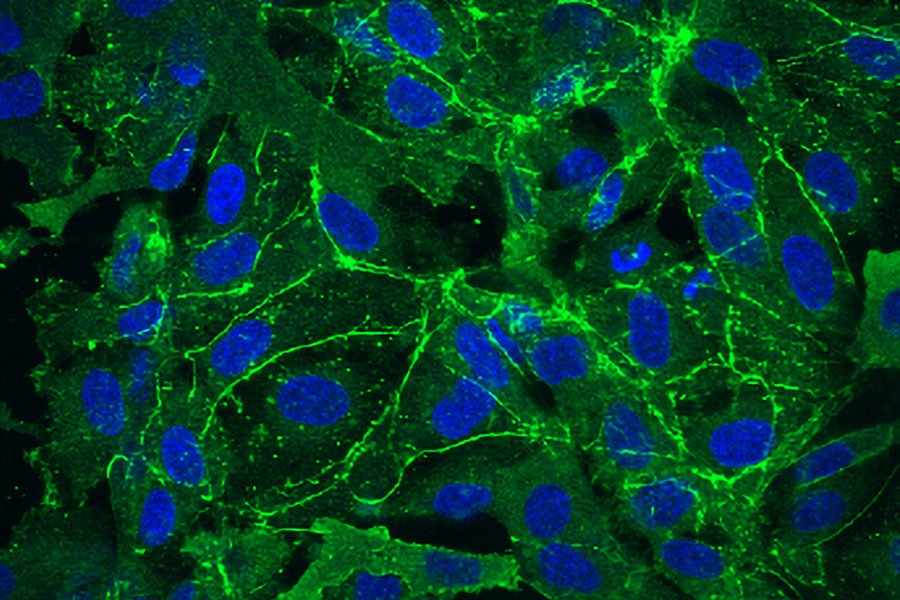
Cientistas do MIT investigaram como as mutações genéticas que causam a síndrome de Rett afetam os vasos sanguíneos do cérebro. As células endoteliais da síndrome de Rett, mostradas aqui, apresentaram menor expressão de ZO-1 (verde), uma proteína essencial para a formação de uma vedação hermética nos vasos sanguíneos, em comparação com as células de controle (não mostradas na imagem). Créditos: Imagem cedida pelos pesquisadores.
Pesquisadores do MIT descobriram que duas mutações genéticas comuns que causam a síndrome de Rett desencadeiam uma cadeia de eventos moleculares que comprometem a integridade estrutural dos vasos sanguíneos em desenvolvimento no cérebro, tornando-os permeáveis. O estudo atribui o problema à superexpressão de um microRNA específico (miRNA-126-3p) e demonstra que a redução dos níveis desse microRNA ajuda a corrigir o defeito vascular.
A síndrome de Rett é um distúrbio grave do desenvolvimento que afeta tanto o cérebro quanto o corpo. É causada por diversas mutações no gene MECP2, amplamente expresso, mas os primeiros sintomas só se tornam aparentes quando as crianças afetadas (principalmente meninas) atingem os 2 ou 3 anos de idade. Como esse é um período crítico no desenvolvimento dos vasos sanguíneos do cérebro, neurocientistas do Instituto Picower para Aprendizagem e Memória do MIT iniciaram um estudo para modelar como duas mutações comuns, porém distintas, do MeCP2 podem afetar o desenvolvimento vascular e contribuir para a profunda patologia neurológica da doença.
Para conduzir a pesquisa publicada recentemente na revista Molecular Psychiatry , o autor principal, Tatsuya Osaki, e a autora sênior, Mriganka Sur, desenvolveram culturas avançadas de tecido humano para modelar o desenvolvimento vascular, com e sem as mutações do gene MeCP2. As culturas não apenas permitiram modelar e observar de perto como as mutações afetavam os vasos, mas também possibilitaram dissecar molecularmente os problemas observados e, em seguida, testar uma intervenção que se mostrou eficaz.
“Já se demonstrou o papel dos microRNAs na síndrome de Rett, mas agora comprovar que o miRNA-126-3p está, na verdade, a jusante do MeCP2 e diretamente implicado na disfunção das células endoteliais é uma peça importante do quebra-cabeça da síndrome de Rett”, afirma Sur, professor titular da Cátedra Newton de Neurociência no Instituto Picower e no Departamento de Ciências Cerebrais e Cognitivas do MIT.
Construção de embarcações e detecção de vazamentos
Com base em anos de experiência em engenharia de tecidos, incluindo um período como pós-doutorando no laboratório do coautor e professor de engenharia mecânica e biológica do MIT, Roger D. Kamm, Osaki construiu “redes microvasculares tridimensionais” usando células-tronco pluripotentes induzidas humanas (células iPS) doadas por pacientes com síndrome de Rett. As células doadas foram induzidas a se tornarem células-tronco e, em seguida, células endoteliais (a estrutura principal dos vasos sanguíneos). Incorporadas em um gel e misturadas com fibroblastos, as células endoteliais se auto-organizaram em redes de tubos, que Osaki então conectou a um sistema microfluídico para fornecer circulação.
Um conjunto de culturas apresentava a mutação R306C. Osaki criou uma microvasculatura de controle geneticamente idêntica, exceto pela ausência da mutação. Outro conjunto de culturas apresentava a mutação R168X. E, novamente, Osaki comparou essa cultura de controle idêntica, exceto pela mutação, utilizando a técnica CRISPR.
A equipe de pesquisa escolheu essas duas mutações porque ambas são relativamente comuns, mas afetam o gene MeCP2 de maneiras diferentes, explica Sur. A descoberta de que cada uma dessas mutações distintas causadoras da síndrome de Rett levou, em última análise, à regulação positiva do miRNA-126-3p e ao comprometimento da integridade dos vasos sanguíneos sugere que os problemas vasculares são, de fato, uma característica central da doença.
“Há algo em comum entre essas mutações”, diz Sur.
Em particular, testes de laboratório mostraram que os vasos sanguíneos com qualquer uma das mutações apresentavam expressão reduzida de uma proteína chamada ZO-1, que é crucial para garantir que as junções entre as células endoteliais nos vasos sanguíneos formem uma vedação hermética (como o rejunte em um piso de azulejos). A ZO-1 também não se localizava nessas junções. De fato, testes adicionais mostraram que as culturas de vasos com a mutação de Rett eram relativamente mais permeáveis em comparação com os controles.
Deficiências semelhantes foram evidentes em outra cultura de células criada pela equipe, na qual adicionaram astrócitos para simular ainda mais de perto a barreira hematoencefálica (BHE), que regula rigorosamente o que pode entrar e sair dos vasos sanguíneos e chegar ao cérebro. Há fortes suspeitas de que problemas na BHE contribuam para doenças neurodegenerativas como Alzheimer , Huntington , ELA e demência frontotemporal .
Para entender melhor como os problemas vasculares podem prejudicar a função neural na síndrome de Rett, os pesquisadores expuseram neurônios ao meio de cultura de vasos sanguíneos de pacientes com Rett. Essas células nervosas apresentaram atividade elétrica reduzida, um possível sinal de que as secreções das células endoteliais da síndrome de Rett afetaram os neurônios.
Capturar um culpado
De modo geral, a função do MeCP2 é reprimir a expressão de outros genes. A expectativa dos cientistas, portanto, era que, quando o MeCP2 fosse comprometido por mutações, o resultado seria a superexpressão de muitos genes. No entanto, o ZO-1 apresentou expressão reduzida. Algo precisava explicar isso, e os miRNAs eram suspeitos, diz Osaki, porque funcionam como reguladores da expressão gênica.
“Por isso, levantamos a hipótese de que deveria haver algum mediador entre a mutação MeCP2 e a regulação negativa de ZO-1 e o aumento da permeabilidade da barreira hematoencefálica”, diz Osaki. “Nos concentramos nos microRNAs.”
De fato, ao analisar o perfil de miRNAs nas culturas de Rett e nos controles, os cientistas descobriram que o miRNA-126-3p estava superexpresso. E, por meio do sequenciamento de RNA, a equipe identificou mais vias moleculares necessárias para manter a integridade vascular, que estavam desreguladas nas culturas de Rett.
Embora o sequenciamento e o perfil associassem a regulação positiva do miRNA-126-3p à alteração da cadeia molecular de eventos, Osaki e Sur buscaram provas mais definitivas. Para obtê-las, trataram as culturas com mutação de Rett com um “antissenso” — uma molécula que reduz os níveis de miRNA-126-3p. Isso resultou em um aumento na expressão de ZO-1 e em uma restauração parcial da função de barreira das células endoteliais — ou seja, menos permeabilidade — nas culturas de vasos. A redução da expressão do miRNA também restaurou as vias moleculares que os cientistas estavam monitorando para estados mais saudáveis.
Descobriu-se que existe um medicamento que inibe o miR-126, chamado miRisten, que está sendo testado clinicamente para o tratamento da leucemia. Osaki e Sur afirmam que planejam administrá-lo a camundongos com síndrome de Rett para verificar se ele apresenta resultados positivos.
Além de Osaki, Sur e Kamm, os coautores do artigo são Zhengpeng Wan, Koji Haratani, Ylliah Jin, Marco Campisi e David Barbie.
O financiamento para o estudo veio de fontes como os Institutos Nacionais de Saúde (NIH), uma bolsa da MURI, a Freedom Together Foundation e o Simons Center for the Social Brain.